Software Validation and Verification under MDR and IVDR
The new European regulations are generating a lot of commotion. And this, what is it due to?
In this article we want to tell you what the new regulations say regarding the validation and verification of software in medical devices under the new MDR/IVDR european regulation.
European regulatory context in medical devices
Harmonized medical device and in vitro standards
The directives/regulations are supported by norms/standards to facilitate their application. The European Commission itself is responsible for making the request to create or update them. Standards made by a standardization body following a request are called harmonized standards. The lists of current harmonized standards for each directive/regulation can be found on the website of the European Commission.
According to the Blue Guide:
“If the references of the harmonized standards have been published in the Official Journal of the European Union, they confer a presumption of conformity with the essential requirements and other legislative requirements that they are intended to cover.”
Also,
“If manufacturers choose not to apply a harmonized standard, they have an obligation to demonstrate that their products meet the essential requirements by other means of their choice (for example, by any existing technical specification, including all other available standards).”
As a conclusion to all of the above, it is good practice to follow the harmonized standards that apply to our product. This way:
- It will be easier to comply with the regulation since the scope of the processes to be carried out is more detailed than in the directive/regulation.
- It will be easier to review the technical documentation by a notified body.
- By being easier to review, it will also be easier to achieve product compliance.
Harmonized standards for software products (year 2020)
If we look for the list of harmonized standards from the directives prior to the new regulations, related to software, we find the following:
- ISO 13485: Quality management system
- ISO 14971: Risk analysis
- IEC 62304: Software life cycle
- IEC 62366-1: Medical devices. Part 1: Application of usability engineering to medical devices.
Harmonized standards for software products (version May 2022)
Ok, and what are the harmonized standards that currently apply?
- ISO 13485: Quality management system
- ISO 14971: Risk analysis
Looking at the previous standards, the reader will wonder how it is possible that there are fewer harmonized standards than in the previous directives.
That could be happening? Why are there fewer harmonized standards than before? Is there a request to update the standards to make them harmonized?
The answer is YES.
Standards required to be harmonized
The following list shows the list of standards that would apply to medical devices that include software:
- IEC 62304: Software life cycle
- IEC 62366-1: Usability
- EN 82304-1: Health software. Part 1: General requirements for product safety
- IEC 81001-5-1: Security and effectiveness of healthcare software and healthcare information technology systems. Part 5-1: Security. Activities in the product life cycle
- IEC TR 60601-4-5: Medical electrical equipment – Part 4-5: Guidance and interpretation – Safety-related technical security specifications.
Currently, the maximum date for adopting these product regulations is May 2024. However, it is likely that this date will be delayed since there is an EU proposal to extend the transition period for the application of the new regulation.
Strategy to follow
We could choose between one of these options:
- Wait for the standards to be updated and appear on the list of harmonized standards?
- Go on applying the current versions of the standards?
The choice depends on the context of each company. Of course, a norm is not applied or internalized in 2 days. Consider the size of the company’s product team to understand how long these implementations may take. Are they new topics or does there already exist, for example, a cybersecurity department?
The suggestion that we give from SQS is to apply the latest versions of the standards and when they appear updated, review the changes and apply them.
One positive point is that cybersecurity regulations, which may be the newest topic in this new regulation, are already published, although they still do not appear on the official list of harmonized standards.
Software validation and verification under new regulation
Definitions validation and verification
There is often confusion between the difference between the validation and verification terms.
Here are the two definitions:
- Definition of Verification: Examination process followed by judgment based on tests that demonstrate that the output elements (processes, documentation, software or applications) of a specific development phase meet the requirements of that phase in terms of completeness. , correctness and coherence.
Example: review of design documents, unit tests
- Validation Definition: Analysis process followed by evidence-based judgment to determine whether an item (for example, a process, documentation, software, or an application) meets a user’s needs, particularly with regard to security and quality, emphasizing the suitability of its operation in accordance with its objective in its intended environment.
Example: system tests
Starting point
In cases where we already have a software product running and we are considering obtaining the CE marking, the following questions are the first to be answered:
- Are we clear that our software is a medical device?
To adequately answer this question, it is necessary to define the intention of use our product.
Once we have defined the intention of use and, therefore, we can affirm that our software is a medical device, we can proceed to ask ourselves the following question:
- Does my product have a risk class established?
Depending on whether the product is a medical or in vitro device, it must be established. Within the regulations there are specific rules to be able to carry out this classification correctly. There are specific MDCG guides to address it even due to its complexity at times.
Verification and Validation Activities
The activities in this regulation are divided according to the risk of the product. The 62304 has the following classification:
- A: No possible injury or damage to health
- B: Non-serious injury possible
- C: Death or serious injury is possible
In this article we focus on validation and verification. Risk management is very important but is outside the scope of validation. Based on some defined risks, requirements are defined to mitigate the risks and these requirements are validated together with the rest of the requirements.
Next we focus on the activities of software development itself. The following table summarizes the activities included in regulation 62304 and which are carried out according to the risk of the medical device:
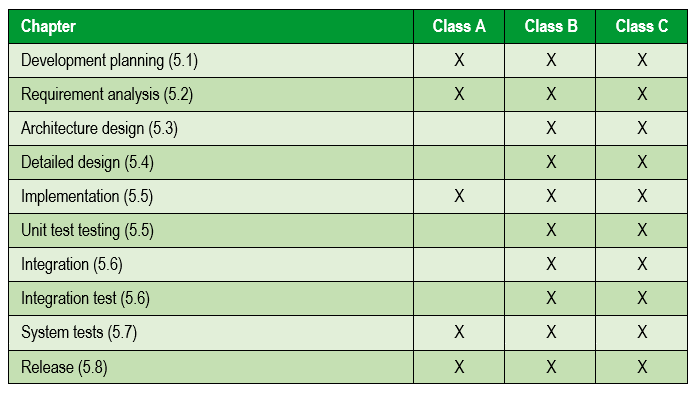
And as a result of all activities, a software validation and verification report is made, which must contain the results of all the previous activities and is included in the following sections:
Software as Medical device validation activities
Among the validation activities that must be carried out, which include both specification and execution and preparation of the test report, are the following:
- Tests for user requirements (82304-1)
Medical device software verification activities
- User requirements (82304-1)
- System requirements (82304-1)
- Software requirements (62304 – 5.2.6)
- Software architecture (62304 – 5.3.6)
- Software Detailed design (62304 – 5.4.4)
Test Result:
- Unit tests (62304 – 5.5.5)
- Integration tests (62304 – 5.6.2)
- Regression tests (62304 – 5.6.6 and 5.6.7)
- Tests for software requirements (62304 – 5.7.1)
- Tests for system requirements (82304-1)
- Test for Cybersecurity (81001-5-1)
Bugs found during testing are usually collected in the following reports:
- Bug report (pending fix)
- Residual Error Report
The software validation and verification report also includes a traceability matrix where the requirements are linked to the results of the test case executions. It shows that there is no untested requirement. If there is any execution with a failed result, it will also be reflected in this matrix.
Summary
At this time we must:
- Periodically review the list of harmonized standards
- Review the publication of standards that are not yet updated
And meanwhile:
- Design and validate our medical device -> based on risk (safety)
- Design and validate our health product -> based on cybersecurity (security)
Conclusion: What is new about the new regulation that affects the design of software medical devices?
The newest without a doubt is the implementation of cybersecurity measures.




Contact us
If you want to know more about the subject or have any other question, do not hesitate, contact us.
Request more information
Certificaciones

ISO 9001

ISO/IEC 27001

ENS-nivel medio

ISO 20000

UNE-EN ISO/IEC 17025
Síguenos
Aviso Legal | Política de Cookies | Contacto
© 2026 Software Quality Systems S.A. | SQS is a member company of Innovalia