Normas Armonizadas Producto Sanitario Software MDR e IVDR
Los nuevos reglamentos europeos MDR (2017/745) y IVDR (7017/746) se apoyan en una serie de normas armonizadas.
En este artículo profundizaremos sobre las normas armonizadas para la obtención del marcado CE de nuestro producto sanitario o in vitro de tipo software.
En el siguiente esquema se muestra un resumen de las normativas que aplican a los dispositivos médicos e in vitro que incluyen software:
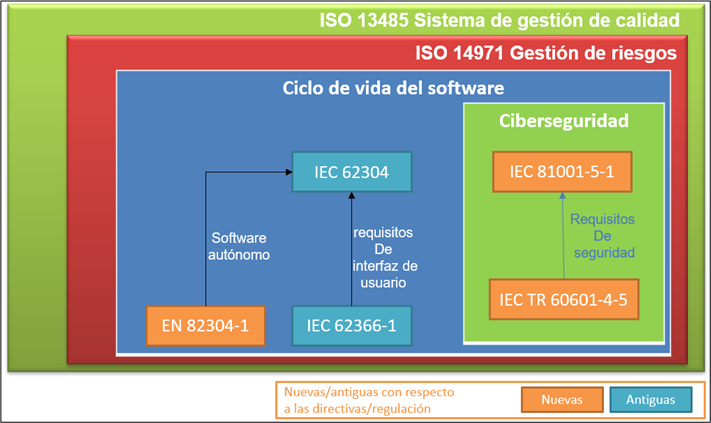
La normativa del sistema de gestión de calidad de los dispositivos médicos es la ISO 13485, la norma de gestión de calidad que debe cumplir una empresa para poder fabricar y distribuir productos sanitarios.
“Visita nuestra página sobre la normativa ISO 13485, sobre el sistema de gestión de calidad y seguridad en dispositivos médicos.”
La norma inmediatamente por debajo y que hace referencia la ISO 13485 es la norma de gestión de riesgos ISO 14971. La gestión de riesgos es fundamental para poder realizar las labores de diseño y desarrollo, y para así luego hacer una buena validación del producto.
“Visita nuestra página sobre la normativa ISO 14971, sobre la Aplicación de Gesstión de Riesgos a los Dispositivos Médicos.”
Bajo el marco de calidad de la ISO13485 y siguiendo la ISO14971 para la gestión de riesgos de nuestro producto, nos encontramos con la normativa IEC 62304, clave para el diseño, desarrollo, validación, verificación y mantenimiento del software, es decir, trata sobre todo el ciclo de vida del software.
“Visita nuestra página sobre la normativa IEC 62304 sobre la aplicación del ciclo de vida del software en dispositivos médicos.”
Otra normativa de aplicación que ya existía en las directivas europeas anteriores era la norma de usabilidad IEC 62366-1. Aplicar esta normativa ayuda a especificar unos requisitos de interfaz de usuario que aseguran que los riesgos asociados a la usabilidad de los dispositivos médicos de tipo software están mitigados.
“Visita nuestra página sobre la normativa IEC 62366-1 sobre la aplicación de la intenigería de la usabilidad a los productos sanitarios.”
Por otro lado y como novedad se encuentra la normativa EN 82304-1 aplicable a software autónomos.
“Visita nuestra página sobre la normativa EN 82304-1 sobre los requisitos generales para la seguridad de los productos sanitarios.”
Y la mayor novedad en este nuevo reglamento es la incorporación de normas armonizadas referentes a ciberseguridad. La normativa IEC 81001-5-1 define el ciclo de vida de desarrollo del software seguro, es decir, añade actividades de diseño, implementación, verificación y validación para asegurar que el software se diseña protegiéndolo de ataques de ciberseguridad.
“Visita nuestra página sobre la normativa IEC 81001-5-1 sobre la implementación de actividades de ciberseguridad al ciclo de vida del software de los productos sanitarios.”
La normativa IEC 81001-5-1 se complementa el technical report IEC TR 60601-4-5 proporcionando los requisitos de seguridad que debe cumplir el producto software.
“Visita nuestra página sobre la normativa IEC TR60601-4-5 sobre las especificaciones técnicas de ciberseguridad aplicada a productos sanitarios.”
Contacta con un experto
Si quieres saber más del tema o tienes cualquier otro tipo de consulta, no lo dudes, ponte en contacto con nosotros.
Servicios Relacionados
Solicita más información
Certificaciones

ISO 9001

ISO/IEC 27001

ENS-nivel medio

ISO 20000

UNE-EN ISO/IEC 17025
Síguenos
Aviso Legal | Política de Cookies | Contacto
© 2026 Software Quality Systems S.A. | SQS is a member company of Innovalia